Médecine fœtale
Publié le 03 mar 2015Lecture 7 min
Trisomie 21 : de l’importance du diagnostic cytogénétique
B. HERVE, D. MOLINA-GOMES, F. VIALARD, Unité fonctionnelle de cytogénéique, Centre hospitalier intercommunal, Poissy-Saint-Germain-en-Laye
Caractérisée par son phénotype particulier au milieu du XIXe siècle, ce n’est que 100 ans plus tard qu’une hypothèse proposant une origine cytogénétique a été formulée, notamment par Raymond Turpin en 1956. Sous son impulsion, Marthe Gautier a mis au point les techniques de culture cellulaire et de préparation cytogénétique, aboutissant à la mise en évidence d’un compte chromosomique à 47 chez les patients dénommés alors «mongoliens» en 1958. C’est à partir de ces préparations que Jérôme Lejeune a identifié le chromosome surnuméraire comme étant un chromosome 21. Le terme de trisomie 21 a, dès lors, été adopté à partir de 1960. Cette découverte marque la naissance de la cytogénétique humaine, ouvrant ainsi la voie à la caractérisation d’autres anomalies chromosomiques.
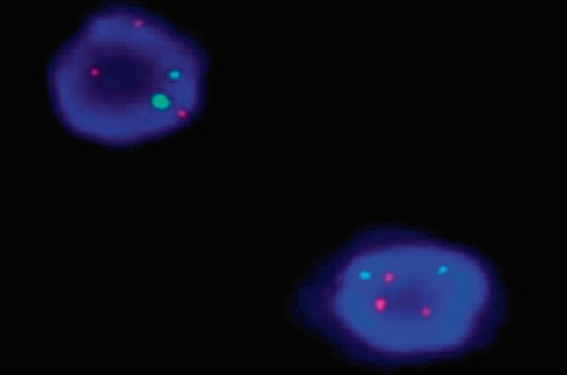
Diagnostic de trisomie 21 et prélèvements fœtaux S’il est une notion fondamentale lorsqu’on aborde le sujet de la trisomie 21, c’est que le diagnostic est et reste cytogénétique. La présence d’un cortège de signes cliniques identifiables en pré et postnatal ne dispense en aucun cas de l’établissement du caryotype. Le caryotype est réalisable à partir de différents types tissulaires. En période prénatale, il s’agit principalement de trophoblaste ou de liquide amniotique, moins fréquemment de sang fœtal. En postnatal, le diagnostic se fait le plus souvent sur sang périphérique et dans de rares cas sur biopsie cutanée. L’examen direct de la biopsie de trophoblaste permet un résultat rapide, en 24 à 48 heures. De résolution limitée, il a avant tout une fonction de «comptage» et permet donc l’identification rapide d’une trisomie 21 (figure 1 a et b). Figure 1 a. Métaphases obtenues par l’examen direct du trophoblaste. Figure 1 b. Caryotype masculin montrant une trisomie 21 libre, issu de l’examen direct du trophoblaste. Les cultures du trophoblaste et du liquide amniotique sont obtenues en 15 à 21 jours en moyenne et sont de meilleure résolution. En pratique, et surtout pour les prélèvements de liquide amniotique, le diagnostic est porté grâce aux techniques dites « de diagnostic rapide » que sont l’hybridation in situ (FISH) sur noyaux interphasiques (figure 2) ou plus rarement la PCR quantitative. Ces techniques permettent un diagnostic en 24 à 72 heures, qui nécessite néanmoins une confirmation par le caryotype après culture cellulaire. Figure 2. FISH sur noyaux interphasiques avec utilisation de sondes spécifiques des chromosomes 13 (vert) et 21 (rouge) montrant la présenec de trois chromosomes 21. Une trisomie 21, des trisomies 21 Bien que l’on parle de la trisomie 21 sous forme d’une seule et même entité clinique, il existe différents types de trisomies 21 d’un point de vue cytogénétique. • La forme de loin la plus fréquente est la trisomie 21 dite « libre et homogène », soit présente dans l’ensemble des cellules soumises à l’analyse cytogénétique. Le caryotype identifie 47chromosomes avec 3 chromosomes 21 indépendants. Elle représente environ 94 % des cas de trisomie 21. • Dans 2 % des cas, il existe une trisomie 21 en mosaïque. Dans ce cas de figure, deux populations cellulaires cohabitent, dans des proportions variables selon les tissus, une population à 47 chromosomes avec un chromosome 21 surnuméraire, et une population normale à 46 chromosomes. Cette forme est le plus souvent due à une erreur mitotique survenue lors des premières divisions cellulaires embryonnaires (soit par non disjonction des chromosomes 21 chez un embryon initialement normal, soit par perte d’un chromosome 21 chez un embryon initialement trisomique). En raison de l’incertitude concernant la localisation des cellules trisomiques, le pronostic reste très réservé et la sévérité du phénotype imprévisible. • Enfin, dans environ 4 % des cas, on observe une trisomie 21 par translocation. Au sein de ce sous groupe, 95 % des cas concernent une translocation robertsonienne c’est-à-dire entre un chromosome 21 et un chromosome acrocentrique (chromosomes 13, 14, 15, 21 et 22). La translocation la plus fréquemment retrouvée est la t(14;21) (figure 3). Figure 3. Caryotype masculin montrant une trisomie 21 par translocation Robertsonienne t(14;21). La translocation t(21;21), deuxième forme rencontrée, d’origine majoritairement de novo, représente un cas particulier au pronostic défavorable si elle est héritée (voir plus loin) (figure 4). Les 5 % de cas restants représentent les trisomies 21 dites «partielles», issues de la transmission de manière déséquilibrée d’une translocation parentale équilibrée entre un chromosome 21 et n’importe quel autre chromosome. Cette situation reste très rarement rencontrée. Figure 4. Caryotype féminin montrant une trisomie 21 par translocation robertsonienne t(21;21). Trisomie 21 et risque de récurrence L’importance de l’établissement du caryotype fœtal prend tout son sens compte tenu du pronostic qu’il implique pour les grossesses ultérieures en fonction du type de trisomie 21 rencontré. En cas de trisomie 21 libre et homogène, il a été constaté que le risque de récurrence est de 2 % (rapport de l’Agence de biomédecine - Diagnostic prénatal 2011(3)). De même, en cas de trisomie 21 en mosaïque, le risque de récurrence théorique semble accru. Différentes hypothèses sont avancées afin d’expliquer ce risque de récurrence augmenté: la malchance, l’âge maternel associé à un vieillissement ovocytaire précoce et à un contrôle moins efficace de la ségrégation chromosomique, l’existence d’une mosaïque germinale auquel s’associe un risque accru d’homotrisomie, et enfin l’existence chez certaines femmes d’une prédisposition à la non-disjonction chromosomique associée à un risque accru d’hétérodisomie. Dans ces deux situations, il est justifié de proposer un diagnostic prénatal pour les grossesses ultérieures sans attendre l’établissement du risque combiné du 1er trimestre. En cas de trisomie 21 par translocation, quelle qu’elle soit, l’établissement du caryotype des parents est nécessaire (figure 5). Figure 5. Arbre décisionnel en cas de trisomie 21 par translocation. Concernant le cas d’une translocation robertsonienne, si celle-ci est de novo, le risque de récurrence est estimé à 2-3 %. Si elle est héritée, le risque théorique dépend du parent porteur : il se situe autour de 10 % s’il s’agit de la mère et autour de 3 % s’il s’agit du père (en raison d’une diminution de la fertilité masculine). Dans le très rare cas de la translocation t(21;21), le risque de récurrence de la trisomie 21 est de 100 % et doit dès lors faire aborder les options de don de gamètes ou d’adoption avec le couple. Dans la pratique courante, il est donc justifié de proposer au couple un diagnostic prénatal pour les grossesses ultérieures, voire d’envisager un diagnostic préimplantatoire. Également, en cas d’implication des chromosomes 14 ou 15 dans la translocation, une recherche de disomie uniparentale est à faire lors pour les prochaines grossesses. Cette prise en charge spécifique est le témoin de l’importance du caryotype pour l’adaptation de la conduite à tenir. De même, concernant le cas particulier de la trisomie 21 partielle, l’établissement du caryotype des parents est indispensable. Là encore, le risque de récurrence théorique est augmenté et doit tenir compte du parent porteur et des chromosomes impliqués. La prise en charge se fait alors au cas par cas. Il est donc dans cette situation également, justifié de proposer un diagnostic prénatal pour les grossesses ultérieures et même d’orienter le couple vers une prise en charge en diagnostic préimplantatoire. La recherche de disomie uniparentale est à considérer selon les chromosomes impliqués (chromosomes soumis à empreinte: 6, 7, 11, 14 et 15). En présence d’une translocation héritée, le conseil génétique doit informer de l’obligation de transmettre ces renseignements aux apparentés (Décret n°2013-527 du 20 juin 2013) pour effectuer une étude familiale, notamment chez les individus en âge de procréer afin de proposer le cas échéant, une prise en charge adaptée.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :